Тон или стиль этой статьи могут не отражать энциклопедический тон, используемый в Википедии . ( Май 2008 г. ) ( Узнайте, как и когда удалить этот шаблон сообщения ) |
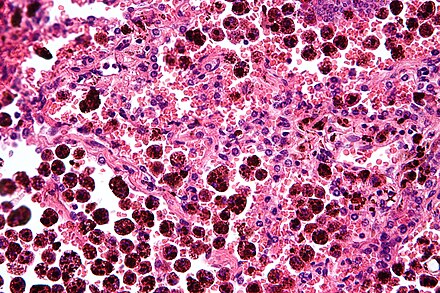
Альвеолярный макрофаг (или пыль клетки ) представляют собой тип макрофага , А профессиональные фагоциты , найдено в легочных альвеолах , у пневмоцитов , но отделена от стенки.
Активность альвеолярных макрофагов относительно высока, поскольку они расположены на одной из основных границ между телом и внешним миром. Они отвечают за удаление частиц, таких как пыль или микроорганизмы, с респираторных поверхностей.
Часто наблюдается, что альвеолярные макрофаги содержат гранулы экзогенного материала, такого как частицы углерода, которые они собрали с респираторных поверхностей. Такие черные гранулы могут быть особенно распространены в легких курильщиков или долгожителей города.
Альвеолярный макрофаг - это третий тип клеток в альвеолах, остальные - пневмоциты I и II типа .
Функция [ править ]

{kind=link}
{kind=link}
{kind=link}

Альвеолярные макрофаги - это фагоциты, которые играют решающую роль в гомеостазе, защите хозяина и ремоделировании тканей. [1] Их плотность населения имеет решающее значение для этих многих процессов. Они очень адаптивны и могут выделять много секретов для взаимодействия с другими клетками и молекулами, используя несколько поверхностных рецепторов . Альвеолярные макрофаги также участвуют в фагоцитозе апоптотических и некротических клеток. [2] Они должны быть селективными по отношению к фагоцитируемому материалу для защиты нормальных клеток и структур. [2] Для борьбы с инфекцией фагоциты способствуют распознаванию множества рецепторов (PRR), помогающих распознавать патоген-ассоциированные молекулярные структуры (PAMP) на поверхности патогенных микроорганизмов. [3]Все PAMP имеют общие черты: они уникальны для группы патогенов, но неизменны по своей базовой структуре; и необходимы для определения патогенности (способности одного организма вызывать инфекционное заболевание в другом организме). [3] Белки, участвующие в распознавании микробного паттерна, включают рецептор маннозы, рецепторы комплемента, DC-SIGN, Toll-подобные рецепторы (TLR), рецептор скавенджера, CD14 и Mac-1. [3] [4] PRR можно разделить на три класса:
- сигнальные PRR, которые активируют механизмы транскрипции генов, которые приводят к клеточной активации,
- эндоцитарные PRR, которые участвуют в связывании патогенов и фагоцитозе, и
- секретируемые PRR, которые обычно действуют как опсонины или активаторы комплемента.
Распознавание и устранение вторгшихся микроорганизмов происходит как опсонин-зависимым, так и опсонин-независимым путями. Молекулярные механизмы, способствующие опсонин-зависимому фагоцитозу, различны для конкретных пар опсонин / рецептор. Например, фагоцитоз опсонизированных IgG патогенов происходит через рецепторы Fcγ (FcγR) и включает в себя удлинения фагоцитов вокруг микроба, что приводит к продукции провоспалительных медиаторов. И наоборот, прием патогена, опосредованного рецептором комплемента, происходит без наблюдаемых расширений мембраны (частицы просто проникают в клетку) и обычно не приводит к медиаторному ответу воспаления.
После интернализации микроб заключен в везикулярную фагосому, которая затем сливается с первичными или вторичными лизосомами, образуя фаголизосому. [3] Существуют различные механизмы, которые приводят к внутриклеточному уничтожению; есть окислительные процессы и другие, независимые от окислительного метаболизма. Первый включает активацию мембранных ферментных систем, которая приводит к стимуляции поглощения кислорода (известному как респираторный взрыв) и его восстановлению до реактивных промежуточных соединений кислорода (ROI), молекулярных форм, которые являются высокотоксичными для микроорганизмов. [3] Фермент, ответственный за возникновение респираторного взрыва, известен как никотинамидадениндинуклеотидфосфат (НАДФН) оксидаза, которая состоит из пяти субъединиц. [3]Один из компонентов представляет собой мембранный цитохром, состоящий из двух белковых субъединиц, gp91phox и p22phox; остальные три компонента являются белками цитозольного происхождения: p40phox, p47phox и p67phox. [3] НАДФН-оксидаза существует в цитозоле AM в состоянии покоя; но после активации два из его цитозольных компонентов, p47phox и p67phox, фосфорилируют свои тирозиновые и сериновые остатки, которые затем могут опосредовать транслокацию NADPHox в компонент цитохрома, gp91phox / p22phox, на плазматической мембране через элементы цитоскелета. [5]
По сравнению с другими фагоцитами респираторный взрыв при AM имеет большую величину. [3] Кислороднезависимые микробицидные механизмы основаны на выработке кислоты, на секреции лизоцимов, на железосвязывающих белках и на синтезе токсичных катионных полипептидов. [3] Макрофаги обладают набором антимикробных молекул, заключенных в их гранулы и лизосомы. [3] Эти органеллы содержат множество деградирующих ферментов и антимикробных пептидов, которые выделяются в фаголизосомы, таких как протеазы, нуклеазы, фосфатазы, эстеразы, липазы и высокоосновные пептиды. [3]Более того, макрофаги обладают рядом механизмов депривации питательных веществ, которые используются для голодания фагоцитированных патогенов по основным микронутриентам. [3] Определенные микроорганизмы разработали контрмеры, которые позволяют им избежать уничтожения фагоцитами. Хотя лизосомальная деградация является эффективным средством нейтрализации инфекции и предотвращения колонизации, некоторые патогены паразитируют на макрофагах, используя их в качестве клетки-хозяина для роста, поддержания и репликации. [3]Такие паразиты, как Toxoplasma gondii и микобактерии, способны предотвращать слияние фагосом с лизосомами, избегая, таким образом, вредного действия лизосомальных гидролаз. Другие избегают лизосом, покидая фагоцитарную вакуоль, чтобы достичь цитозольного матрикса, где их развитие не затруднено. В этих случаях макрофаги могут быть задействованы для активного уничтожения фагоцитированных микроорганизмов, производя ряд высокотоксичных молекул и вызывая депривационный механизм для их голодания. [3] Наконец, у некоторых микробов есть ферменты для детоксикации метаболитов кислорода, образующихся во время респираторного взрыва. [3]
Когда их недостаточно для отражения угрозы, альвеолярные макрофаги могут выделять провоспалительные цитокины и хемокины, вызывая высокоразвитую сеть защитных фагоцитарных клеток, ответственных за адаптивный иммунный ответ.
Легкие особенно чувствительны и склонны к повреждению, поэтому, чтобы избежать побочного повреждения пневмоцитов 1 и 2 типа, альвеолярные макрофаги находятся в состоянии покоя, вырабатывая мало воспалительных цитокинов и проявляя небольшую фагоцитарную активность, о чем свидетельствует подавленная экспрессия фагоцитов. рецепторный антиген макрофага 1 (Mac-1). [1] [6]АМ активно подавляют индукцию двух систем иммунитета организма: адаптивного иммунитета и гуморального иммунитета. Адаптивный иммунитет подавляется воздействием AM на интерстициальные дендритные клетки, B-клетки и T-клетки, поскольку эти клетки менее избирательны в отношении того, что они разрушают, и часто вызывают ненужное повреждение нормальных клеток. Чтобы предотвратить неконтролируемое воспаление в нижних дыхательных путях, альвеолярные макрофаги выделяют оксид азота, простагландины, интерлейкины-4 и -10 (IL-4, IL-10) и трансформирующий фактор роста-β (TGF-β). [6] [7] [8] [9]
Оксид азота [ править ]
NO является основным источником иммуномодуляции у грызунов и вырабатывается ферментом синтетазой оксида азота типа 2 (NOS2) в альвеолярных макрофагах. [8] NO ингибирует фосфорилирование тирозина киназ, участвующих в выработке рецептора интерлейкина-2 (IL-2), экспрессия которого является фундаментальной для пролиферации Т-клеток. [7] Однако у людей активность NOS2 проверить трудно. [8]
Существует два объяснения отсутствия реакции промотора индуцибельной синтетазы оксида азота (iNOS) человека на активацию NO липополисахаридами (ЛПС) + интерферон гамма (IFNγ). [8] Во-первых, существуют различные варианты инактивирующих нуклеотидов в человеческом аналоге энхансерного элемента, который регулирует индуцированную LPS / IFNγ экспрессию гена NOS2 мыши. Во-вторых, из-за отсутствия ядерного фактора в макрофагах человека, который необходим для оптимальной экспрессии гена NOS2 (комплекс LPS-индуцируемый ядерный фактор-каппа B / Rel). [8] Предполагается, что сложность верификации NOS2 связана с гораздо более жестко контролируемой экспрессией в AM человека по сравнению с экспрессией в AM грызунов. [8]NOS2 является частью ауторегуляторной петли обратной связи, в которой аллерген или провокатор стимулирует выработку воспалительных цитокинов, что, в свою очередь, стимулирует выработку NO, а NO подавляет выработку цитокинов. [8] У крыс NO ингибирует опосредованное гранулоцитарно-макрофагальным колониестимулирующим фактором (GM-CSF) созревание дендритных клеток, а у людей он ингибирует TNF-альфа-опосредованное созревание дендритных клеток человека посредством циклической GMP-зависимой механизмы. [8] NO продлевает способность дендритных клеток человека усваивать антигены в очагах воспаления, модулируя тем самым начальные шаги, ведущие к антиген-специфическим иммунным ответам. [8]
Считается, что продукция NO имеет отношение к патологии астмы. У пациентов с астмой наблюдается повышенная экспрессия iNOS в эпителиальных клетках дыхательных путей и повышенный уровень оксида азота в выдыхаемом воздухе. [8]
Эндопероксид простагландина 2 (PGE2) [ править ]
Выделено множество других иммуномодулирующих факторов, наиболее важными из которых являются простагландины и цитокины. PGE2 был первым описанным иммуномодулятором, полученным из макрофагов. [8] PGE2 участвует в усилении транскрипции IL-10 лимфоцитами периферической крови и продукции белка; а также в дезактивации макрофагов и Т-клеток. [8] PGE2 представляет собой иммуномодулирующий эйкозаноид, полученный из компонента клеточной мембраны, арахидоновой кислоты , и обрабатывается в каскаде арахидоновой кислоты: последовательной оксигенации и изомеризации арахидоновой кислоты ферментами циклооксигеназы и PGE2-синтазы. [10] Регулирование клеток-мишеней с помощью PGE2 происходит посредством передачи сигналов через четыре связанных с клеточной мембраной G-белок-связанных рецепторов E-простаноидов (EP), названных EP1, EP2, EP3 и EP4. [10] PGE2 ингибирует уничтожение бактерий и продукцию ROI с помощью AM за счет нарушения Fcγ-опосредованного фагоцитоза за счет своей способности стимулировать продукцию внутриклеточных эффекторов циклического аденозинмонофосфата (цАМФ) посредством передачи сигналов рецепторов EP2 и EP4. [5] [10] Рецепторы EP2 и EP4 передают сигнал в основном через стимулирующий G-белок (Gs), повышая активность аденилатциклазы (AC) и последующее образование цАМФ. [5]цАМФ является вторым мессенджером, который влияет на множественные клеточные функции посредством активации двух последующих эффекторных молекул, протеинкиназы А (ПКА) и обменных белков, непосредственно активируемых цАМФ (Epac-1 и -2). [5] Epac-1 и PKA являются важными факторами, участвующими в ингибировании уничтожения AM бактерий. [5] Эффект PKA является результатом его способности фосфорилировать остатки серина и треонина на многие клеточные белки, особенно на белок, связывающий элемент ответа цАМФ фактора транскрипции (CREB). Ось цАМФ / PKA / CREB опосредует ингибирование высвобождения TNF-альфа. [5] Уничтожение фагоцитированных бактерий AM зависит от нескольких различных микробицидных механизмов, таких как снижение опосредованного НАДФН-оксидазой высвобождения ROI. [3][5] Создание ROI под действием НАДФН-оксидазы является важным бактерицидным механизмом после FcR-опосредованного фагоцитоза. [5] PGE2 активирует рецепторы EP2 и EP4, связанные с Gs, путем лигирования, стимулирования продукции цАМФ и последующей активации последующих эффекторов цАМФ, PKA и Epac-1; оба, в свою очередь, нарушают фосфорилирование и транслокацию через фагосомную мембрану компонента НАДФН-оксидазы, p47phox, тем самым ингибируя респираторный взрыв. [5]
Интерлейкин-4 и -10 [ править ]
IL-4 - это плейотропный цитокин, который играет ключевую роль в развитии Т-хелперных клеток 2 типа (Th2). IL-4 важен для дифференцировки наивных CD4-T-клеток в зрелые клетки типа Th2; а также для переключения класса иммуноглобулинов (Ig) на IgE и IgG4 во время развития иммунных ответов. [11] [12] Ig - это класс антител, обнаруженных только у млекопитающих, которые играют важную роль в аллергической реакции и защите от многих видов патогенов, защищая организм от них путем активации комплемента, опсонизации для фагоцитоза и нейтрализации их токсинов. . [12]
Было показано, что как IL-4, так и IL-10 снижают продукцию металлопротеиназ (эндопептидаз, расщепляющих коллаген и другие внеклеточные белки) AM человека. [8] [9] ИЛ-4 оказывает двойное действие на биологическую функцию макрофагов, которое может быть либо стимулирующим, либо ингибирующим. [9] Он усиливает экспрессию антигена MHC класса II (внеклеточный белковый комплекс, который взаимодействует исключительно с CD4-T-клетками как часть экзогенного пути) и Mac-1 (поверхностный рецептор как часть врожденной системы комплемента), тем самым способствуя фагоцитозу. [9] Было также показано, что IL-4 ингибирует выработку PGE2 за счет снижения экспрессии фермента простагландин H-синтазы-2 (PGHS-2), который имеет решающее значение в производстве PGE2. [8]Однако ИЛ-4 подавляет выработку ФНО-альфа, ИЛ-1 и -6, которые являются важными цитокинами в провоспалительном ответе). [9]
IL-10 подавляет секрецию провоспалительных цитокинов TNF-альфа и INF-гамма, подавляя тем самым пролиферацию Т-клеток, NK-клеток и AM. [8] ИЛ-10 имеет аналогичные иммуномодулирующие механизмы с TGF-β. [8] Считается, что оба цитокина снижают скорость апоптоза в альвеолярных макрофагах человека, таким образом косвенно усиливая опосредованное альвеолярными макрофагами ингибирование пролиферации Т-клеток. [8] При активации бактериальными продуктами происходит значительное увеличение базальной скорости апоптоза. Апоптоз, в частности, регулируется наличием цитокинов: IFNγ увеличивает скорость апоптоза, тогда как IL-10 и TGF-β снижают ее. [8]Однако IL-10 оказывает контрпродуктивное действие на иммунную систему, и было показано, что он действительно способствует заражению чужеродными патогенами. Роль IL-10 в бактериальной и паразитарной инфекции была обнаружена как стратегия уклонения от иммунной системы хозяина. [13] Существуют бактерии, которые паразитируют на AM, проникая через их мембраны, и процветают, растя и размножаясь внутри них, используя AM как клетки-хозяева. Обычно эту инфекцию можно устранить с помощью Т-клеток, которые активируют ферменты в альвеолярных макрофагах, которые уничтожают бактерии; но было показано, что эти бактерии изменяют сигнальную сеть цитокинов в свою пользу. В качестве ингибирующего цитокина IL-10 способствует инфицированию альвеолярных макрофагов и моноцитов человека, полностью обращая защитный эффект IFNγ против внутриклеточной репликации Legionella pneumophila. [13]Yersinia enterocolitica также высвобождает антиген вирулентности LcrV, который индуцирует IL-10 через Toll-подобный рецептор-2 и CD14 (дополнительный поверхностный белок TLR4-опосредованной передачи сигналов LPS), что приводит к подавлению IFNγ и TNF-alpha. подавление. [13]
Трансформирующий фактор роста β (TGF-β) [ править ]
В нормальных условиях альвеолярные макрофаги плотно прилегают к альвеолярным эпителиальным клеткам, тем самым вызывая экспрессию интегрина αvβ6. Интегрины представляют собой димерные рецепторы на поверхности клетки, состоящие из альфа- и бета-субъединиц, которые активируют TGF-β. < [14] [15] TGF-β - это многофункциональный цитокин, который модулирует различные биологические процессы, такие как рост клеток, апоптоз, внеклеточный матрикс синтез, воспаление и иммунные ответы. [16] TGF-β строго регулирует противовоспалительную активность, подавляя выработку провоспалительных цитокинов, тем самым подавляя функцию Т-лимфоцитов. [17]Интегрины avβ6 и avβ8 изолируют латентный TGF-β на поверхности клетки, где активация может быть тесно связана с клеточными ответами на стресс окружающей среды при поддержании гомеостаза; интегрины также локализуют активированный TGFβ вблизи макрофагов. [18] Обычно зрелый TGFβ секретируется в виде латентного комплекса с его N-концевым фрагментом, латентно-ассоциированным пептидом (LAP), который ингибирует его активность. [16] Латентный комплекс ковалентно связан с внеклеточным матриксом путем связывания с латентными TGF-β-связывающими белками. [14] TGF-β активируется различными механизмами в легких, в конечном итоге включающими протеолиз или конформационное изменение LAP. [18]Интегрин αvβ6 способен опосредовать активацию TGF-β за счет связывания с LAP TGF-β1, который служит сайтом связывания лиганда для интегрина и является важным компонентом аппарата активации TGF-β. [16] [19] После активации TGFβ приводит к подавлению функциональности макрофагов (выработка цитокинов и фагоцитоз). [16] Связывание активированного TGF-β с его рецепторами, экспрессируемыми на альвеолярных макрофагах, вызывает нижестоящий сигнальный каскад, включая фосфорилирование рецепторно-регулируемых гомологов 2 и 3 декапентаплегических (R-SMAD) малых матерей (R-SMAD). [1] [16] [17 ] ]Фосфорилированные SMAD-2 и -3 затем образуют гетеромерные комплексы с общим медиатором SMAD 4 (co-SMAD-4). После сборки комплексы перемещаются в ядро через ядерные поры с помощью импортинов альфа / бета. Попадая в ядро, эти комплексы накапливаются и в конечном итоге действуют как факторы транскрипции, регулируя экспрессию генов-мишеней TGF-β. [17] Таким образом, передача сигналов TGF-β включает прямой путь от рецепторов на поверхности клетки к ядру.
Активация [ править ]
Toll-подобные рецепторы (TLR) представляют собой сигнальные PRR , способные распознавать различные бактериальные белки. [4] Хотя бактерии разработали способы уклонения от защитных механизмов хозяина, они экспрессируют PAMP, такие как липогликаны и липопротеины, которые распознаются клетками врожденной иммунной системы через TLR. [4] При связывании PAMP с TLR, TLR запускает воспалительные и защитные реакции в клетке-хозяине, вызывая полимеризацию актина в альвеолярных макрофагах (критический компонент в эндоцитозе и подвижности). [16]Полимеризация актина в альвеолярных макрофагах вызывает подавление экспрессии интегрина, что, в свою очередь, вызывает дезактивацию TGF-β и подавление базального уровня фосфорилирования SMAD 2/3; впоследствии приводя к активации и отделению альвеолярных макрофагов от альвеолярных эпителиальных клеток [16] [15]. После активации макрофаги становятся готовыми к фагоцитозу и начинают секретировать провоспалительные цитокины (TNF-α и IL-6). [16]
Праймирование макрофагов включает усиление активности респираторного взрыва IFN-γ и TNF-α. [3] IFNγ индуцирует как повышенное сродство НАДФН-оксидазы к НАДФН в макрофагах, так и повышенную скорость транскрипции генов и экспрессию сообщений для белка gp91phox. [3] TNF-α действует как аутокринный стимул, увеличивая экспрессию транскриптов p47phox и p67phox. Области интереса, полученные во время респираторной вспышки, в свою очередь, увеличивают выработку TNF-α макрофагами. [3]
Деактивация [ править ]
Газообмен должен быть восстановлен как можно быстрее, чтобы избежать побочного повреждения, поэтому активированные лимфоциты секретируют IFNγ, чтобы стимулировать выработку матриксной металлопротеиназы MMP-9 макрофагами. [16] Сообщается, что AM продуцируют MMP-9 частично через PGE2-зависимые пути передачи сигналов PKA, которые являются путями, участвующими в ингибировании фагоцитоза. [20] MMP-9 активирует латентный TGF-β, повторно индуцируя экспрессию интегринов αvβ6 на альвеолярных эпителиальных клетках, тем самым возвращая альвеолярный макрофаг в состояние покоя. [1] [16] [20] Активация TGF-β также полезна, потому что его продукция стимулирует синтез коллагена в интерстициальных фибробластах, который необходим для восстановления архитектуры альвеолярной стенки. [16]
Сравнение пигментированных альвеолярных макрофагов [ править ]
| Болезнь | Макрофаги | Внешний вид пигмента (пятно HE) | Связанная история болезни | Изображение | Комментарий к изображению |
|---|---|---|---|---|---|
| Антракоз | Черно-коричневые гранулы |
| Черная стрелка показывает интерстициальный антракотический пигмент. Можно предположить, что близлежащие макрофаги (белая стрелка) содержат антракотический пигмент. В легких случаях у пожилых людей отчет не является обязательным. | ||
| Респираторный бронхиолит | «Макрофаги курильщика» | От желтого до светло-коричневого, мелкозернистый [23] | Курение табака | Макрофаги курильщика (стрелка); мягкий интерстициальный лимфоцитарный инфильтрат и легкий фиброз (стрелка) [24] | |
| Хроническая легочная гиперемия | Сидерофаги | Коричнево-золотистый и преломляющий. [25] |
| Сидерофаг (черная стрелка) и интерстиций с отеком, отложением гемосидерина (черная стрелка) и коллагеновым утолщением. |
См. Также [ править ]
- Список типов клеток человека, полученных из зародышевых листков
Ссылки [ править ]
- ^ a b c d Lambrecht, BN (апрель 2006 г.). « « Альвеолярный макрофаг на водительском сиденье » ». Иммунитет . 24 (4): 366–8. DOI : 10.1016 / j.immuni.2006.03.008 . PMID 16618595 .
- ^ a b Guyton AC (2007). «Глава 33: Физиология дыхательной системы». Учебник медицинской физиологии . С. 431–433.
- ^ a b c d e f g h i j k l m n o p q r s Stafford JL, Neumann NF, Belosevic M (2002). «Макрофаги-опосредованная врожденная защита хозяина от простейших паразитов». Критические обзоры в микробиологии . 28 (3): 187–248. DOI : 10.1080 / 1040-840291046731 . PMID 12385499 . S2CID 38166749 .
- ^ a b c Круцик С.Р., Модлин Р.Л. (февраль 2004 г.). «Роль Toll-подобных рецепторов в борьбе с микобактериями». Семинары по иммунологии . 16 (1): 35–41. DOI : 10.1016 / j.smim.2003.10.005 . PMID 14751762 .
- ^ a b c d e f g h i Серезани Ч., Чанг Дж., Баллинджер М. Н., Мур Б. Б., Аронофф Д. М., Петерс-Голден М. (ноябрь 2007 г.). «Простагландин E2 подавляет уничтожение бактерий в альвеолярных макрофагах, ингибируя НАДФН-оксидазу» . Американский журнал респираторной клетки и молекулярной биологии . 37 (5): 562–70. DOI : 10,1165 / rcmb.2007-0153OC . PMC 2048683 . PMID 17585108 .
- ^ a b Холт П.Г., Оливер Дж., Билык Н., МакМенамин С., МакМенамин П.Г., Краал Г., Тепен Т. (февраль 1993 г.). «Подавление функции (й) антигенпредставляющих клеток легочных дендритных клеток in vivo резидентными альвеолярными макрофагами» . Журнал экспериментальной медицины . 177 (2): 397–407. DOI : 10,1084 / jem.177.2.397 . PMC 2190916 . PMID 8426110 .
- ^ a b Bunn HJ, Hewitt CR, Grigg J (май 2002 г.). «Подавление пролиферации аутологичных мононуклеарных клеток периферической крови альвеолярными макрофагами от маленьких детей» . Клиническая и экспериментальная иммунология . 128 (2): 313–7. DOI : 10.1046 / j.1365-2249.2002.01848.x . PMC 1906398 . PMID 12041510 .
- ^ a b c d e f g h i j k l m n o p q r Бингиссер Р. М., Холт П. Г. (апрель 2001 г.). «Иммуномодулирующие механизмы в нижних дыхательных путях: опосредованные оксидом азота взаимодействия между альвеолярными макрофагами, эпителиальными клетками и Т-клетками» . Швейцарский медицинский еженедельник . 131 (13–14): 171–9. PMID 11345807 .
- ^ a b c d e Lacraz S, Nicod L, Galve-de Rochemonteix B, Baumberger C, Dayer JM, Welgus HG (август 1992). «Подавление биосинтеза металлопротеиназы в альвеолярных макрофагах человека интерлейкином-4» . Журнал клинических исследований . 90 (2): 382–8. DOI : 10.1172 / JCI115872 . PMC 443112 . PMID 1322938 .
- ^ a b c Брок Т.Г., Серезани С.Х., Карстенс Дж.К., Петерс-Голден М, Аронофф Д.М. (январь 2008 г.). «Влияние простагландина E2 на субклеточную локализацию белков Epac-1 и Rap1 во время фагоцитоза, опосредованного рецептором Fcgamma, в альвеолярных макрофагах» . Экспериментальные исследования клеток . 314 (2): 255–63. DOI : 10.1016 / j.yexcr.2007.10.011 . PMC 2390918 . PMID 18021770 .
- ^ Pouliot Р, Turmel В, Gelinas Е, Laviolette М, Биссоннетт Е.Ю. (июнь 2005 г.). «Продукция интерлейкина-4 альвеолярными макрофагами человека». Клиническая и экспериментальная аллергия . 35 (6): 804–10. DOI : 10.1111 / j.1365-2222.2005.02246.x . PMID 15969673 . S2CID 22847451 .
- ^ a b Пол УЕ (май 1991 г.). «Интерлейкин-4: прототип иммунорегуляторного лимфокина» . Кровь . 77 (9): 1859–70. DOI : 10.1182 / blood.V77.9.1859.1859 . PMID 2018830 .
- ^ Б с Yoshizawa S, Tateda K, T, Matsumoto Gondaira F, S, Miyazaki Стандифорд TJ, Yamaguchi K (май 2005 г.). «Legionella pneumophila уклоняется от опосредованного гамма-интерфероном подавления роста за счет индукции интерлейкина-10 в макрофагах, полученных из костного мозга» . Инфекция и иммунитет . 73 (5): 2709–17. DOI : 10.1128 / IAI.73.5.2709-2717.2005 . PMC 1087334 . PMID 15845473 .
- ^ a b Арая Дж, Камбье С., Моррис А., Финкбайнер В., Нишимура С.Л. (август 2006 г.). «Интегрин-опосредованная активация трансформирующего фактора роста-бета регулирует гомеостаз легочной эпителиально-мезенхимальной трофической единицы» . Американский журнал патологии . 169 (2): 405–15. DOI : 10,2353 / ajpath.2006.060049 . PMC 1698780 . PMID 16877343 .
- ↑ Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, Glick A, Sheppard D (март 2003 г.). «Потеря опосредованной интегрином альфа (v) бета6 активации TGF-бета вызывает Mmp12-зависимую эмфизему». Природа . 422 (6928): 169–73. DOI : 10,1038 / природа01413 . PMID 12634787 . S2CID 4407206 .
- ^ a b c d e f g h i j k Takabayshi K, Corr M, Hayashi T., Redecke V, Beck L, Guiney D, Sheppard D, Raz E (апрель 2006 г.). «Индукция гомеостатической цепи в легочной ткани микробными соединениями» . Иммунитет . 24 (4): 475–87. DOI : 10.1016 / j.immuni.2006.02.008 . PMID 16618605 .
- ^ Б с Ray CA, Lasbury ME, Durant PJ, Ван SH, Zhang C, Liao CP, Tschang D, Ли CH (2006). «Трансформирующая активация фактора роста-бета и передача сигналов в альвеолярной среде при пневмоцистной пневмонии». Журнал эукариотической микробиологии . 53 Дополнение 1: S127–9. DOI : 10.1111 / j.1550-7408.2006.00200.x . PMID 17169028 . S2CID 37439751 .
- ^ a b Эннес Дж. П., Мангер Дж. С., Рифкин Д. Б. (январь 2003 г.). «Осмысление скрытой активации TGFbeta» . Журнал клеточной науки . 116 (Pt 2): 217–24. DOI : 10,1242 / jcs.00229 . PMID 12482908 .
- ^ Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D (февраль 1999 г.). «Интегрин альфа v бета 6 связывает и активирует латентный TGF бета 1: механизм регуляции легочного воспаления и фиброза». Cell . 96 (3): 319–28. DOI : 10.1016 / S0092-8674 (00) 80545-0 . PMID 10025398 .
- ^ a b Ohbayashi H, Shimokata K (апрель 2005 г.). «Матричная металлопротеиназа-9 и ремоделирование дыхательных путей при астме». Текущие цели в отношении лекарств. Воспаление и аллергия . 4 (2): 177–81. DOI : 10.2174 / 1568010053586246 . PMID 15853739 .
- ^ Котран; Кумар, Коллинз (1999). Патологическая основа болезни Роббинса . Филадельфия: Компания WB Saunders. ISBN 978-0-7216-7335-6.
- ↑ Морган В.К. (ноябрь 1978 г.). «Промышленный бронхит» . Br J Ind Med . 35 (4): 285–91. PMC 1008445 . PMID 367424 .
- ^ Уильям Перри, доктор медицины, магистр здравоохранения, Кристина Конопка, доктор медицины "Легкие - другой интерстициальный пневмонит / фиброз - респираторный бронхиолит" .CS1 maint: multiple names: authors list (link) Тема завершена: 1 июля 2020 г. Незначительные изменения: 1 июля 2020 г.
- ^ Соуза, Селия; Родригес, Марсиу; Карвалью, Андре; Виамонте, Барбара; Кунья, Руи; Гимарайнш, Сусана; де Моура, Консейсан Соуто; Мораиш, Антониу; Перейра, Хосе Мигель (2019). «Диффузные заболевания легких, связанные с курением: выводы из радиолого-патологической корреляции» . Взгляд на визуализацию . 10 (1). DOI : 10.1186 / s13244-019-0765-Z . ISSN 1869-4101 .
- Эта статья распространяется на условиях Международной лицензии Creative Commons Attribution 4.0 ( http://creativecommons.org/licenses/by/4.0/ ) - ^ Зандер, Дэни (2018). Легочная патология . Филадельфия, Пенсильвания: Эльзевир. ISBN 978-0-323-39308-9. OCLC 968711140 . - VII Острая травма легких с сидерофагами
Внешние ссылки [ править ]
- Гистологическое изображение: 13906loa - Система обучения гистологии в Бостонском университете - «Дыхательная система: легкие (человека), альвеолярные макрофаги»
- Гистология в КУМК респ -респ16 «Альвеолы».
- Слайд на ufl.edu